
AI, protein folding, and beyond
Published:
A deep dive into the attention mechanism, AlphaFold2, ESM-2, and de novo protein design
The protein folding problem
The so-called central dogma of molecular biology states (in its most basic form) that genetic information flows from DNA (genes), to RNA (in the form of messenger RNA), to proteins.
Proteins are of the utmost importance for life, as they form most of the enzymes in charge of catalyzing chemical reactions, and play a major structural role serving as scaffolds for many cellular components. They are linear chains of amino acids that fold into intricate three-dimensional structures. This three-dimensional structure dictates to a very high degree the function of the protein. Despite this, until recently our ability to predict the three-dimensional structure of a protein from its amino acid sequence was quite precarious, and even nowadays having tools that can predict such structure with a high-degree of accuracy, our understanding of the folding process remains partially misunderstood. We are however aware of some factors playing an important role in such process:
Amino acid residues form hydrogen bonds with each others, giving rise to structures known as α-helices and β-sheets.
Close-range interactions (i.e., Van der Waals) contribute to the folding process as some residues are tightly packed.
As any other polymer, the structure tends to adopt preferred backbone bond orientations.
Some amino acids are electrically charged, which make them repel or attract to other residues.
Within living organisms, proteins find themselves in an aqueous environment and thereby hydrophobic residues tend to aggregate towards the core, while polar ones tend to do so towards the surface.
Ultimately this process is ruled by thermodynamics, and as such stable shapes must exist in a deep minimum within an energy landscape. [1] Even then, this is a simplified version of the phenomenon, as proteins are not completely rigid structures, as evidenced by intrinsically disordered proteins (IDPs), which do not have a definite three-dimensional structure. [2] Moreover, even if we had a perfect statistical thermodynamics-based simulator, we would not be able to predict the structure of some “rigid” proteins with it alone, as some of them need the help of other proteins (referred to as chaperones) to correctly fold. [3]
Classical approaches for three-dimensional structure prediction
The main two classical approaches used to tackle the protein folding problem have either been physics-based or homology-based.
The former uses forcefields to model the evolution of folding subjected to the forces mentioned in the last subsection. This models are extremely computationally burdensome, and have only been successfully applied to small proteins. Also, the formulation of such forcefields does not represent a completely accurate depiction of the real phenomenon as simplifications are introduced to keep the simulations tractable.
The latter approach relies on the immense catalogue of three-dimensional structures experimentally obtained by researchers over the years using techniques like X-ray crystallography or cryogenic transmission electron microscopy. This structures are routinely deposited in databases like the Protein Data Bank (PDB). Homology-based approaches work under the assumption that similar amino acid sequences fold in similar ways. When the sequence of an unknown protein is presented, similar sub-sequences are looked up in the database and are predicted to fold in the same manner. This process is repeated until the full structure is recovered.
Since 1994 the Critical Assessment of protein Structure Prediction (CASP) has been held with the aims of improving predictions. Before the recent advent of deep learning (DL)-based methods, the homology-based approach saw much more success than the physics-based one. [1]
State of the art approaches to the protein folding problem
Since DeepMind’s introduction of AlphaFold during CASP13 in 2018, the field has been dominated by DL. This first iteration relied on a convolutional neural network (CNN) with residual connections, trained on more than 29,000 PDB structures. The network was able to predict pairwise distances between all amino acid residues of the protein as well as torsion angles between residues from pairwise features derived from a multiple sequence alignment (MSA), which is created by aligning a sequence of interest with similar ones (homologues), showing then differences among them (i.e., amino acid deletions, insertions or substitutions). The idea is that residues that evolve together, are likely to contact each other in the three-dimensional structure. The initial distances and angles outputted by the network are then used to construct a potential, which is minimized using gradient descend in order to obtain a more accurate prediction of the structure. [4]
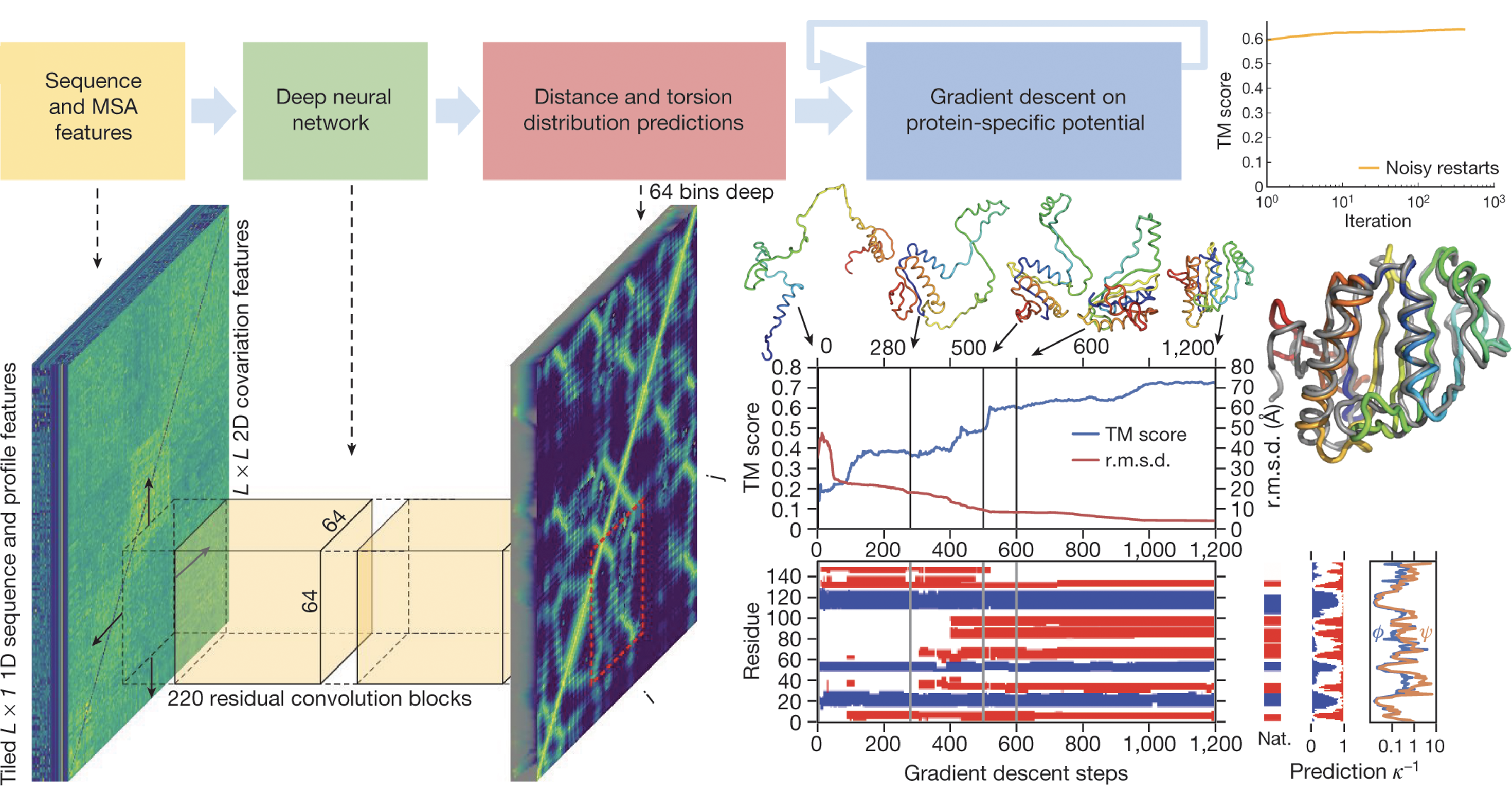
While the results achieved by the first iteration of AlphaFold were certainly a step up from classical approaches, as evidenced by them obtaining the best overall results during CASP13, for CASP14 DeepMind presented yet another model; AlphaFold2. [5] This second version of AlphaFold is based on the the transformer architecture introduced by Google in 2017, in a now famous paper addressing language translation called “Attention is all you need”. [6]The key innovation of the architecture is the incorporation of an attention mechanism, which allows the model to focus on different parts of an input sequence. Attention involves three types of vectors: queries, keys, and values. Each token (i.e., a numerical representation of a word, sub-word, punctuation mark, etc.) in the input sequence broadcasts one of these vectors. A query q (of size dK, for an N-token input) represents how much attention the token being currently processed should pay to other tokens in the input data. Keys represent all elements in the input data, and are used along with the query to assess their relevance to each other. All keys can be collected into a key matrix K of size N × dK. The multiplication of q and K generates a matrix of weights that then needs to be further multiplied by a vector of values V, of size N × dV, which itself contains the actual (unweighted) information the model will pass to further layers. The multiplication of q and K is scaled by $\sqrt{d_K}$ so that each row has unit variance, and a softmax is applied to convert it to a probability distribution where each row sums to 1. To better understand the concept consider the following example, where W matrices are weights learned during training, and x vectors are the embedding (continous, high-dimensional) reprentations of the tokens (words in this case):
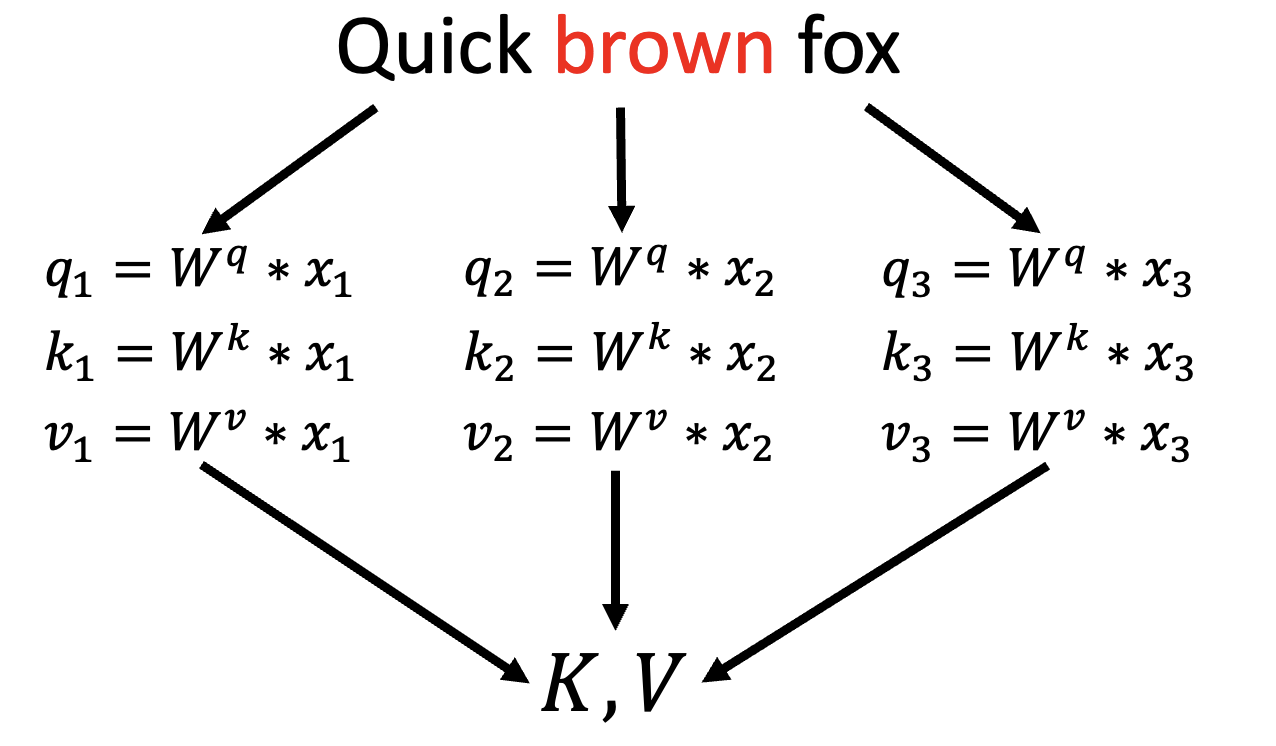
The attention for the word brown can be calculated in the following manner:
$$\text{Attention}_{\text{brown}}(\mathbf{q_2}, \mathbf{K}, \mathbf{V}) = \text{softmax}\left(\frac{\mathbf{q_2K}^T}{\sqrt{d_K}}\right)\mathbf{V}$$ More generally, the attention for the whole sentence can be calulated all at once by collecting all queries into a query matrix Q.
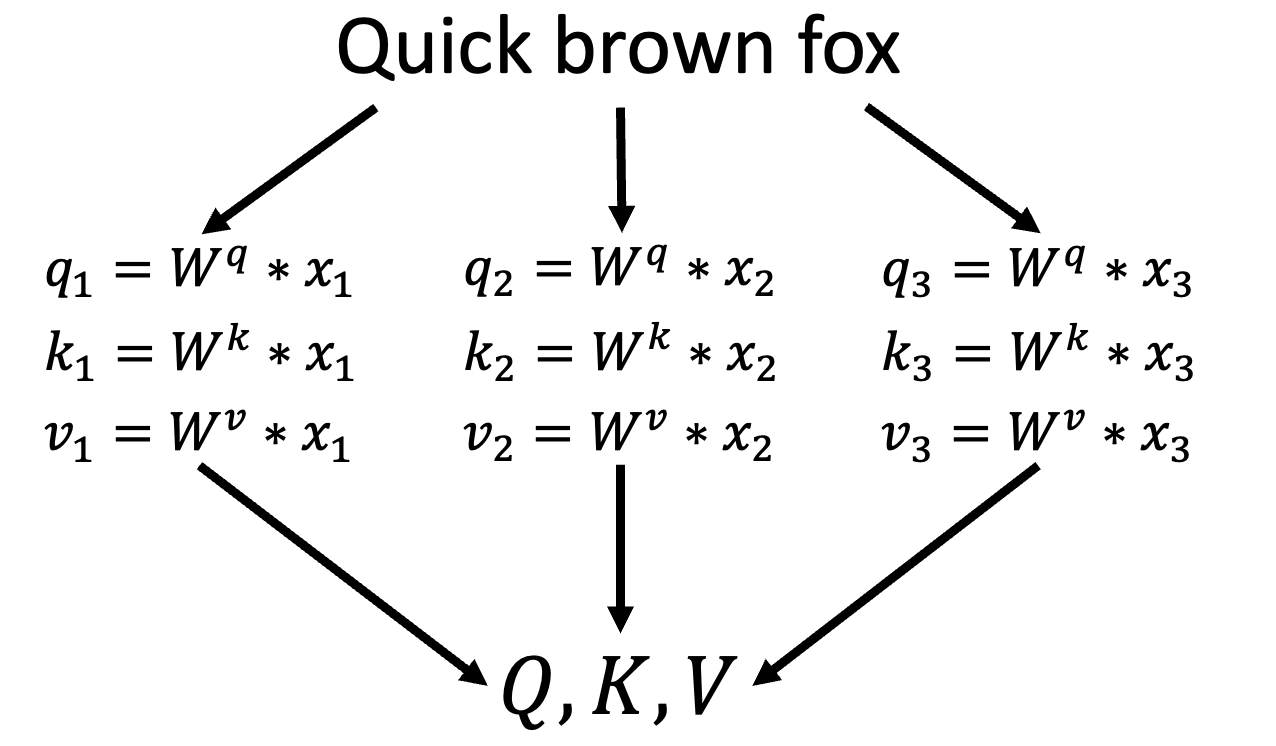
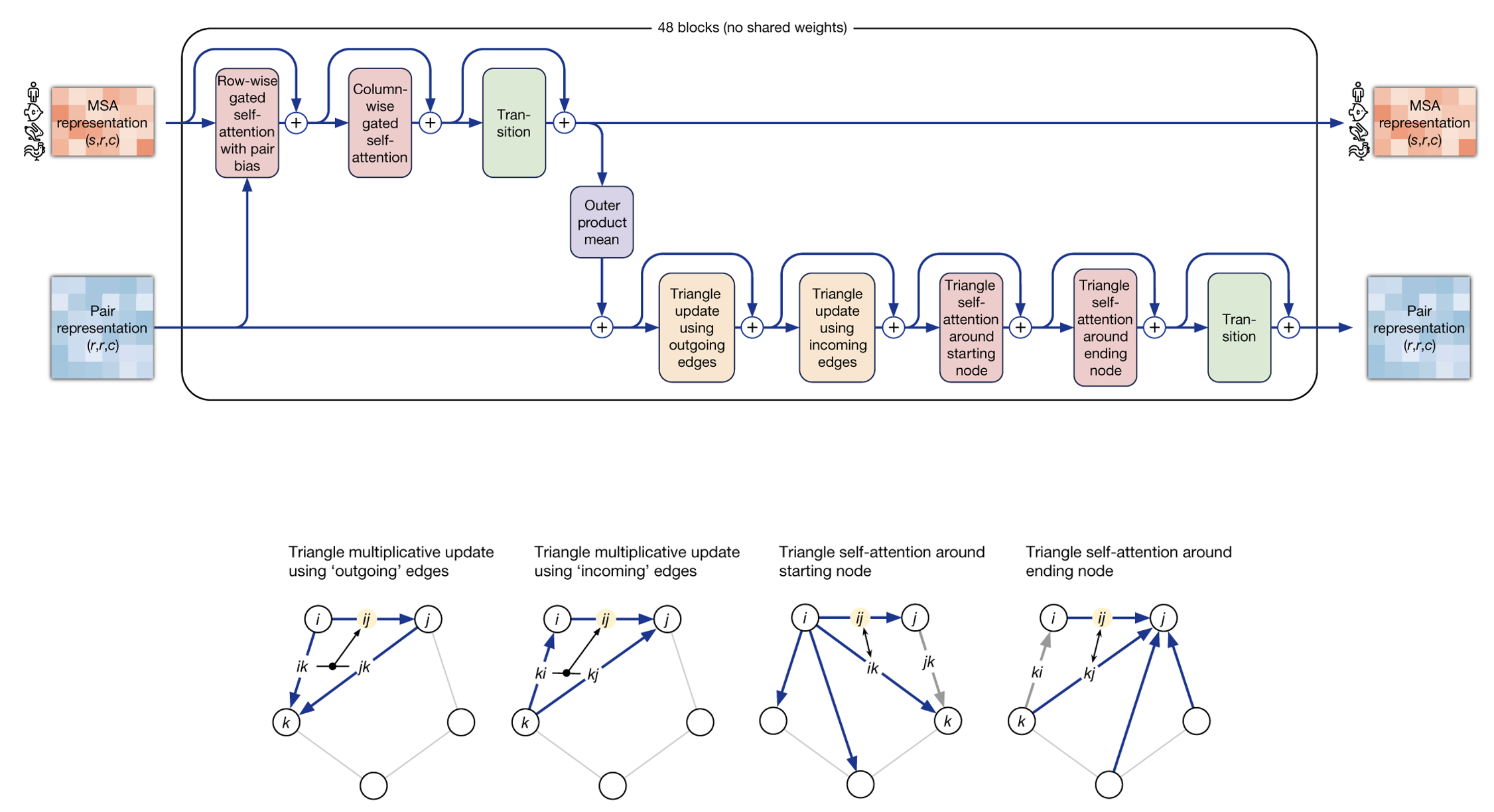
$$\text{Attention}(\mathbf{Q}, \mathbf{K}, \mathbf{V}) = \text{softmax}\left(\frac{\mathbf{QK}^T}{\sqrt{d_K}}\right)\mathbf{V}$$ Attention allowed this version of AlphaFold to overcome the most obvious drawback of its predecessor; the fact that it could only process a limited number contiguous amino acid residues (equal to the convolution kernel size; 64 in this case) at a time. AlphaFold2 introduced attention within “evoformer” blocks. This blocks both take as input and produce a MSA and a pairwise map (again, a sort of initial guess of residue positions in space) representations.
At this point it is worth dedicating a few words regarding how the MSA representation is created from a raw MSA. This process is very simmilar to the “Quick brown fox” example above. Basically the sequences are tokenized by assigning a unique integer to each of the 20 aminoacids, one to gaps, and another for “unknown”. After this, the tokens are embedded into higher-dimensional space and passed trough the network as usual.
The first evoformer block in the network takes the initial MSA and a pairwise map representations and produces updated versions of them, which are then fed to a subsequent block and so on. Within the evoformer block both row-wise attention and then column-wise attention are calculated for the MSA representation. Row-wise attention would find relationships within the same protein homologue, while column-wise attention would do so for equivalent positions among different homologues. Additionaly, some constraints (learned during training) are imposed for the structure to be physically plausible. For example, considering the residues as nodes, the triangle inequality is imposed for all possible combinations of three distinct nodes: $$ d(i,j) \leq d(i,k) + d(j,k) $$ Where d(x,y) represents the distance between residues x and y. This is done by updating distances from every i to every j using “triangular” operations involving a third residue k.
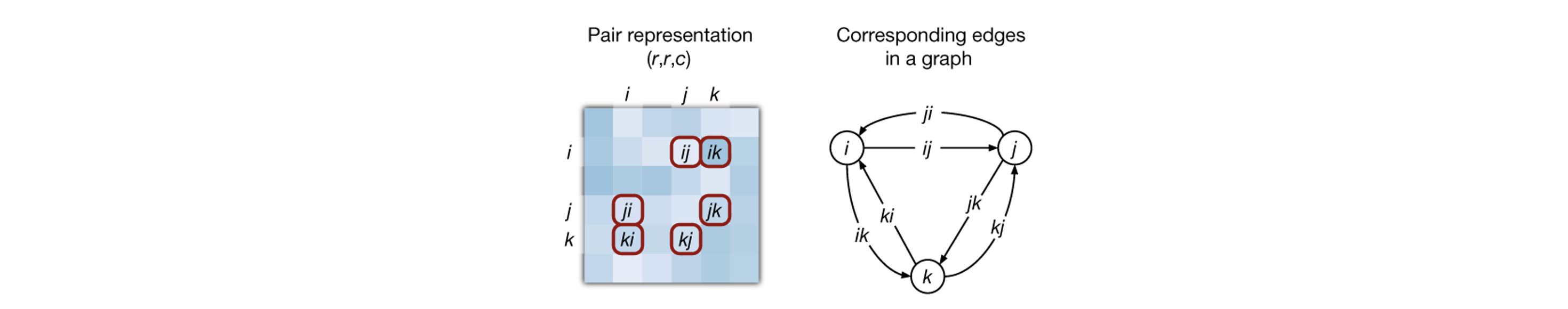
To be a bit more precise, the attention used by this module for the rows is sligtly different than that presented before due to the introduction of an addtional bias term. This is a very important detail as the bias is derived from each amino acid pair within the pairwise map (i.e., \( b_{ij} \)), and it is the mechanism by which it “leaks” information onto the MSA representation for its update. Adding a bias term to the column-wise attention on the other hand would not make any sense, as columns do not represent a single protein structure, but rather equivalent positions of different protein homologues. The attention between residues ij (ignoring scaling), would then be:
$$\text{Row-Wise Attention}_{\text{ij}}(\mathbf{q_i}, \mathbf{k_j}, \mathbf{v_j}, \mathbf{b_{ij}}) = \text{softmax}\left(\mathbf{q_i}^T \mathbf{k_j} + \mathbf{b_{ij}}\right) \mathbf{v_j}$$ Other relevant operations include the calculation of the outer product between colum pairs of the MSA representation to update the pairwise representation, a couple “triangular multiplicative updates”, in which each residue in the pairwaise representation is updated based on its two neighbors using simple multiplication operations, and “triangular self-attention updates” in which the update to a residue pair ij around i is calculated using a query derived from ij, a key and value derived from ik, and a bias derived from jk
$$\text{Triangular Attention}_{\text{ij}}(\mathbf{q_{ij}}, \mathbf{k_{ik}}, \mathbf{v_{ik}}, \mathbf{b_{jk}}) = \text{softmax}\left(\mathbf{q_{ij}}^T \mathbf{k_{ik}} + \mathbf{b_{jk}}\right) \mathbf{v_{ik}}$$ followed by an equivalent update around j (that is, Triangular Attentionji).
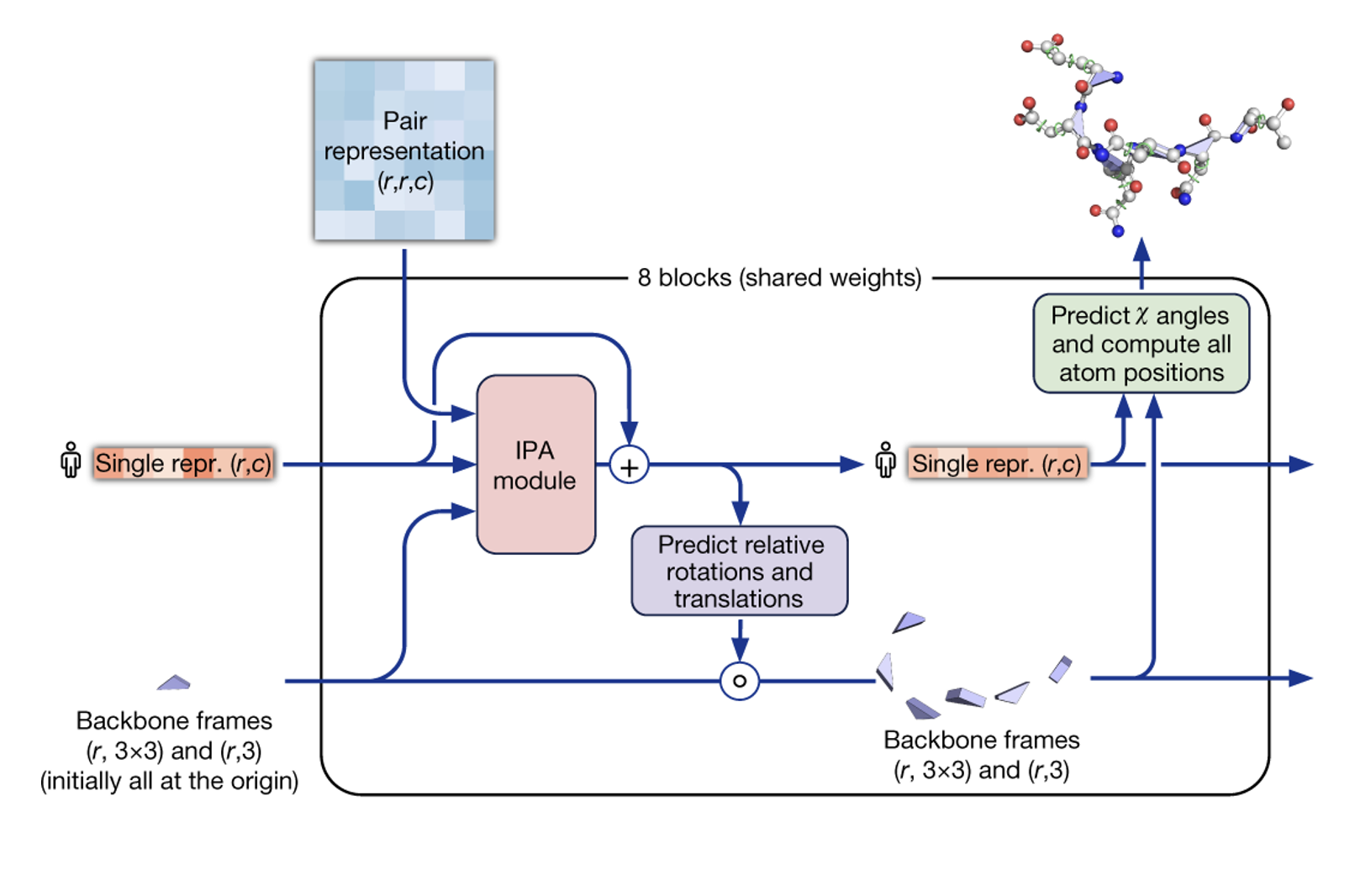
After the evoformer blocks, “structure module” blocks are introduced, whose main function is to compute an invariant point attention (IPA; not to be confused with the beer). The IPA mechanism in these blocks is designed to process the spatial relationships between amino acids in a protein while maintaining invariance to the overall positioning and orientation of the protein structure. It does this by focusing on the relative positions and orientations of amino acids in their local frames of reference, rather than their absolute positions in 3D space. This means that the output of the IPA is not affected by how the protein is rotated or translated globally, making it consistent regardless of the protein’s orientation.
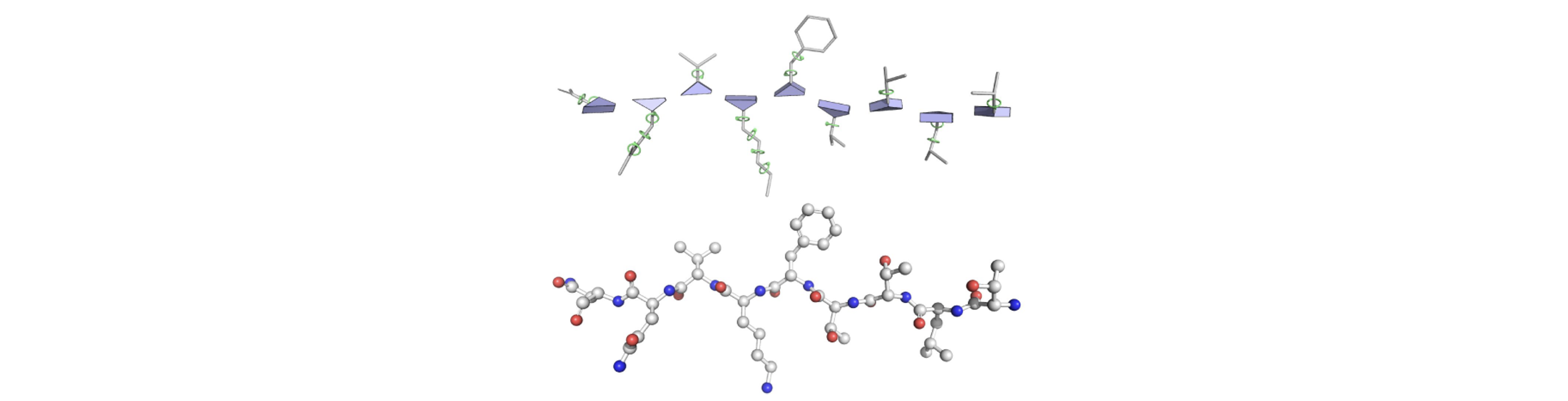
To understand how this is done, we need to introduce the concept of a "residue gas". A protein is a chain of linked amino acids, each composed of three backbone atoms (always a \( N \), \( C^{\alpha} \), and \( C' \)), and a side which varies per specific amino acid type. The idea of AlphaFold is to construct one triangle per residue whose vertices correspond to \( N \), \( C^{\alpha} \), and \( C' \), and detach each residue from its neighbors. As the residues are now detached, they are allowed to move freely (hence the name residue gas). Each residue is parameterized in Euclidean space using a translation vector (\( t_{i} \), the distance from the origin to the \( C^{\alpha} \) carbon), and a rotation matrix (\( R_{i} \)), as \( T_{i} = (t_{i}, R_{i}) \).
At first, all residues are initialized at the origin and they are free to move during inference. It is the job of the structure module, and in particular of the IPA, to update the position these triangles with the help of the pairwise representation coming from the evoformer.
At first it might seem odd to go trough all this trouble since the pairwise map represents the predicted distances between residues, but the nice thing about this residue gas representation is that it additionally accounts for angles, which are neccessary to construct the protein structure prediction.
IPA is quite simmilar to the attentions we have already encountered acconting for one-dimensional relationships, but it additionally introduces a Euclidean-aware term (presented here in a slightly simplified way for the sake of clarity):
$$ \text{IPA}_{\text{ij}}(\mathbf{q_i}, \mathbf{k_j}, \mathbf{v_j}, \mathbf{b_{ij}}, \mathbf{T_i}, \mathbf{T_j}) = \text{softmax}\left(\mathbf{q_i}^T \mathbf{k_j} + \mathbf{b_{ij}} + \| \mathbf{T_i q_i - T_j k_j} \|^{2}\right) \mathbf{v_j}$$ Altough in principle it is posible for residues contiguos in the linked linear sequence to end up far appart in three-dimensional space, in practice this is unlikely as it is heavily penalized in the loss function.
After this is done, some subtle adjusments are preformed in a “classical” manner to comply with idealized bond lenghts and angles for both the backbone and side chains.
A total of three forward passes trough the network are performed to refine the prediction.
As in the first version, once the three-dimensional structure is predicted, a relaxation of the structure is performed using gradient descent on a force field. [5] All in all the picture looks like this:
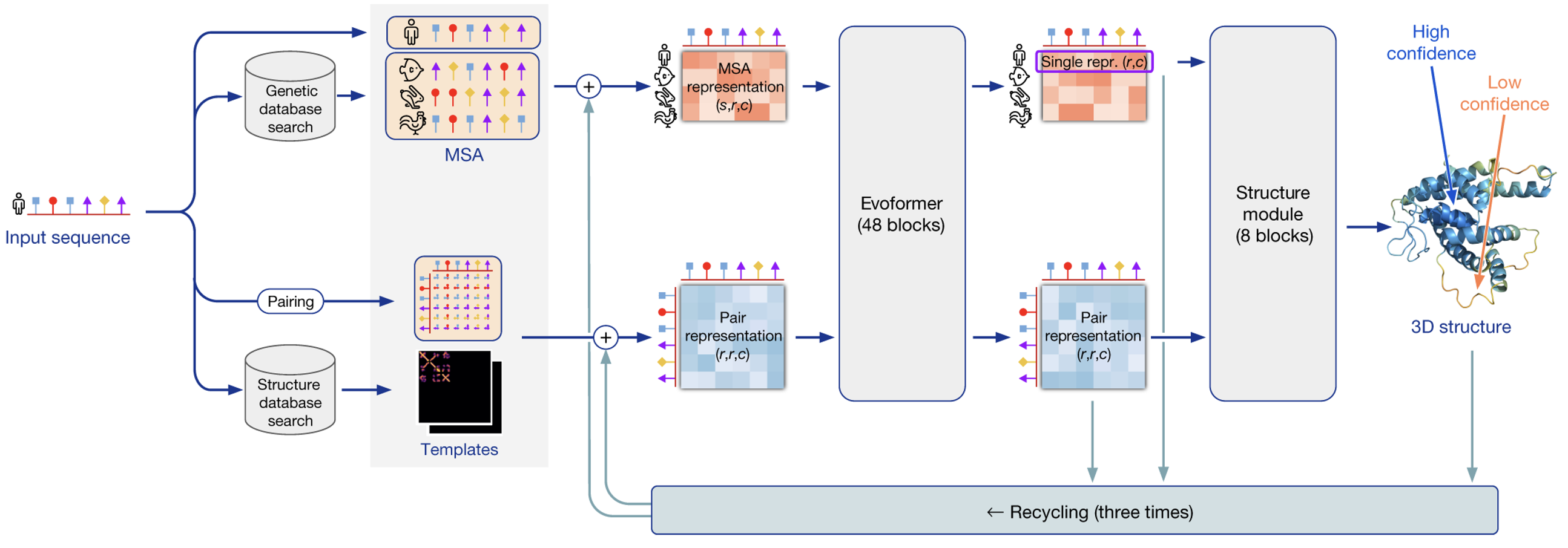
There are of course further details and more precise explanations regarding the implementation in [5]'s supplementary information, but this comprises a fairly complete insight of the inner workings of AlphaFold2.
With these innovations, DeepMind managed to become again the best performing team at CASP. The results however were on this occasion even more outstanding, to the extent that some people considered the protein folding problem to be effectively solved.
DeepMind did not present any model to CASP15, but many teams adapted AlphaFold2 to obtain modest improvements for particular circumstances. [7]
What is next?
Although AlphaFold2 was certainly a breakthrough, challenges still remain. For example, the original version of AlphaFold2 did not account for protein complexes. Some researchers started “hacking” AlphaFold by connecting different proteins forming part of a complex with “flexible linkers” (i.e., chains polar amino acid residues like glycine and serine that serve to connect two main structures, but have close to no impact in the folding of such main structures) to overcome the single-sequence constraint of the model. In 2022 however, DeepMind introduced AlphaFold-multimer to address this issue. [8] While this has work great for some complexes, it still performs poorly for some others, notably for antibody-antigen complexes, which are of special importance for the medical field. [7] IDPs remain a big challenge, and models that address the inherent lack of rigidity in some proteins are needed to accurately solve the protein folding problem. [2] In my opinion, new models should address this issue by not only providing the predicted most stable structure but rather the set of most likely conformations a protein could take over time. Of course, at present, this is a very difficult problem to address, mostly due to the general lack of data regarding structural dynamics.
A field very intimately linked with protein folding still in its infancy is that of protein design. Some approaches based on DL have seen some success in generating custom proteins. For example, one based on a type of network called “message-passing neural network” is able to generate a protein sequence to match a desired shape with reasonable accuracy. [9]
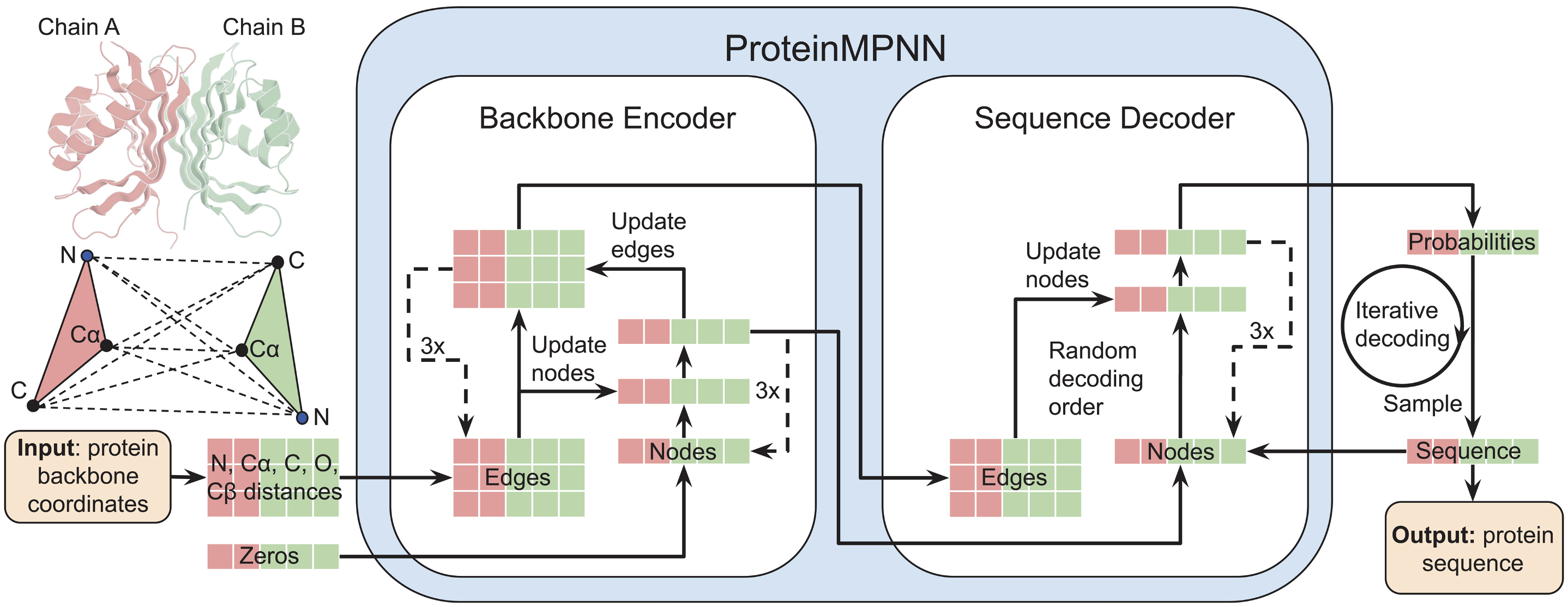
Other researchers have taken the large language model (LLM) approach and used a generative decoder-only transformer (similar to GPT models). This model is prompted with a tag that represents the desired functionality of a protein, and generates one amino acid at a time until the protein is completed. [10]
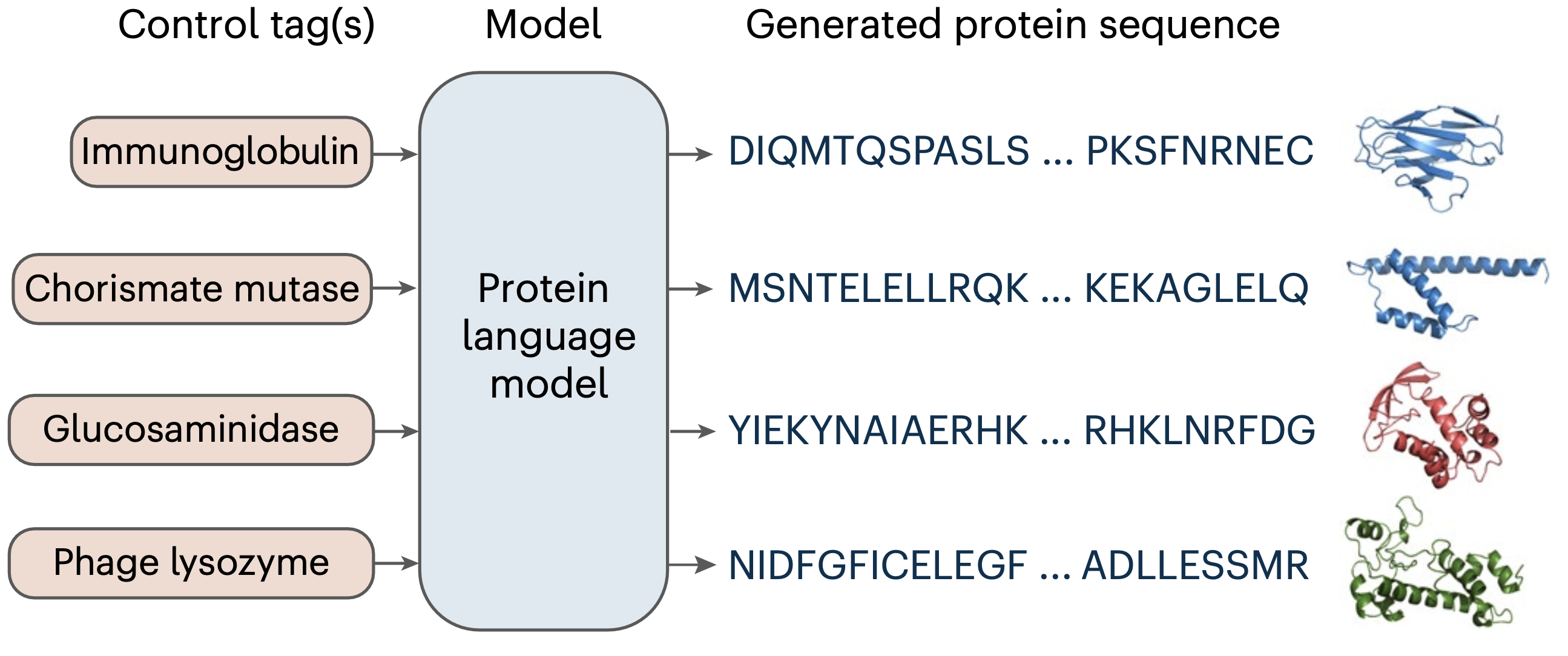
As the field evolves and models become more effective, they will likely be prompted with natural language, describing more complex desired functions rather than prompting using only tags like “Immunoglobulin” or “Phage lysozyme”. As it stands now, this model only explicitly accounts for the linear protein sequence. Other than a LLM module however, these sort of models will probably include an in-line three-dimensional structure predictor (AlphaFold2-based or otherwise). Making explicit use of a three-dimensional representation would likely increase the chances generated sequences are actually functional.
On a similar note, researchers at Meta have adopted an innovative approach to address the protein folding problem. Instead of using features derived from a MSA, they have trained an LLM (coined evolutionary scale modeling-2; ESM-2) to learn general patterns regarding “the language of proteins” in an unsupervised manner. They did this by masking some amino acids from linear sequences, and tasking ESM-2 with predicting those masked residues. For predicting three-dimensional structures, they plug ESM-2 into an AlphaFold-like model using modified evoformer blocks (called “folding trunk” blocks) followed by structure blocks identical to those found in AlphaFold2. This unified model is called ESMFold, and was further trained in a supervised manner using three-dimensional structures from the PDB and predicted by AlphaFold2.
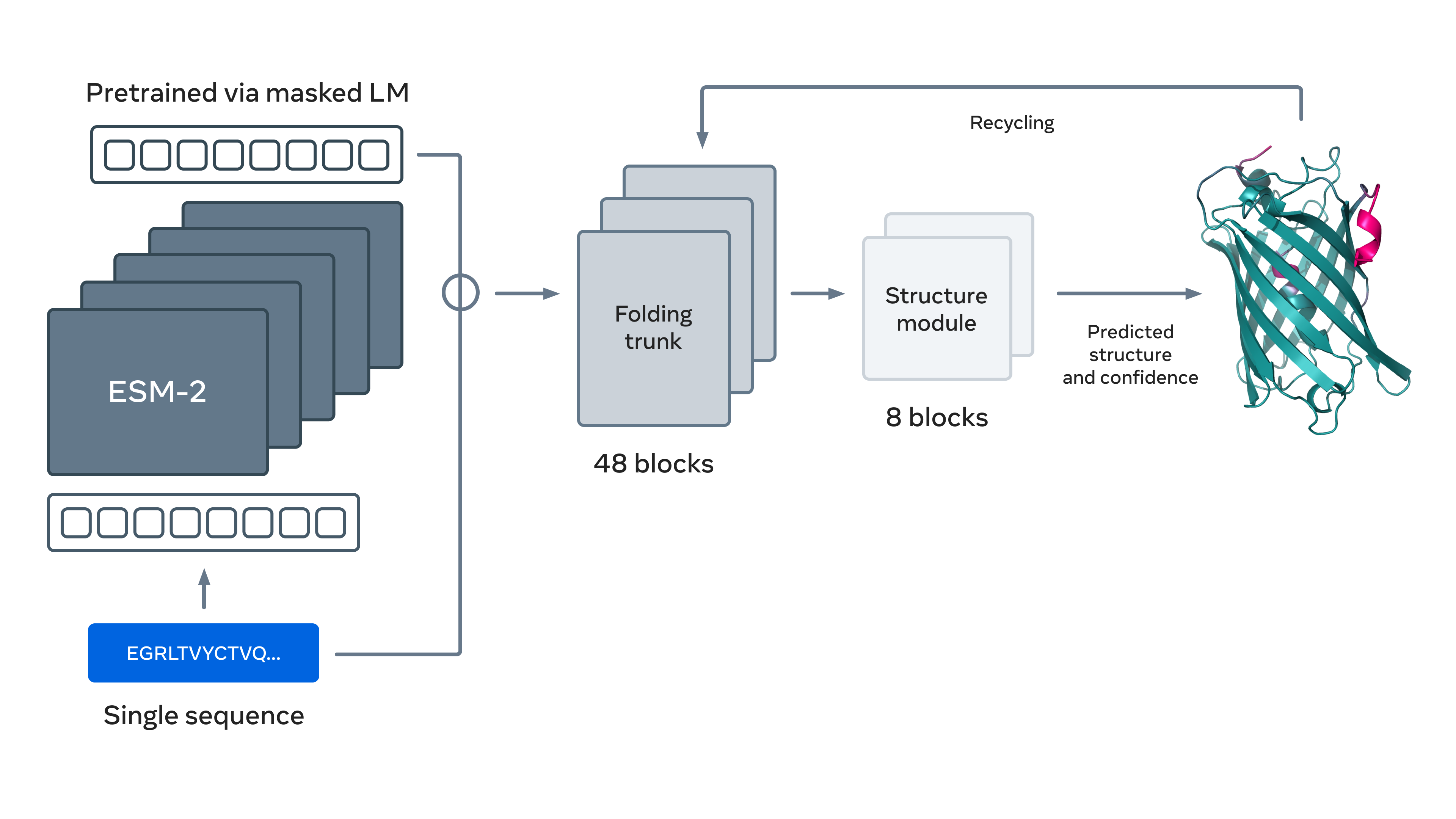
The model is able to perform inference about an order of magnitude faster than AlphaFold2 (mainly because only a linear sequence is required rather than a MSA), albeit taking a hit regarding the quality of the predictions. Regardless, what is remarkable about ESMFold is its ability to predict three-dimensional shapes without being trained using MSAs, suggesting ESM-2 learned some intricate general patterns of this protein language. ESM-2 or similar models could then potentially be harnessed to perform a plethora of different tasks, including protein design. [11]
Conclusion
The introduction of AlphaFold models represented a pivotal moment in the field of protein folding prediction. These DL models have significantly outperformed traditional approaches in predicting protein structures, paving the way for breakthroughs in related fields like drug discovery and molecular biology.
However, several challenges remain unresolved. Accurately modeling protein complexes and IDPs are among the critical areas needing further research and development. Also, since these models are trained on previously seen proteins, predictions on proteins containing exotic, never-before-seen folds might be inaccurate. The future of this field lies in the refinement of these models to capture the dynamic nature of proteins, and in integrating advanced methods from the field of natural language processing to obtain more accurate and innovative models for protein design.
References
1. Dill, K. A., MacCallum, J. L.: The Protein-Folding Problem, 50 Years On. Science 338, 1042–1046 (2012).2. Ruff, K. M., Pappu, R. V.: AlphaFold and Implications for Intrinsically Disordered Proteins. Journal of Molecular Biology 433(20), Academic Press, 167208 (2021). https://doi.org/10.1016/j.jmb.2021.167208
3. Kim, Y. E., Hipp, M. S., Bracher, A., Hayer-Hartl, M., Hartl, F. U.:Molecular chaperone functions in protein folding and proteostasis. Annual Review of Biochemistry 82, 323–355 (2013). https://doi.org/10.1146/annurev-biochem-060208-092442
4. Senior, A. W. et al.: Improved protein structure prediction using potentials from deep learning. Nature 577(7792), 706–710 (2020). https://doi.org/10.1038/s41586-019-1923-7
5. Jumper, J. et al.: Highly accurate protein structure prediction with AlphaFold. Nature 596(7873), 583–589 (2021). https://doi.org/10.1038/s41586-021-03819-2
6. Vaswani, A. et al.: Attention Is All You Need. ArXiv (2017).
7. Callaway, E.: Protein-folding contest seeks next big breakthrough. Nature 613 (2023).
8. Evans, R. et al.: Protein complex prediction with AlphaFold-Multimer. (2022). https://doi.org/10.1101/2021.10.04.463034
9. Dauparas, J. et al.: Robust deep learning-based protein sequence design using ProteinMPNN. Science (1979) 378, 49–56 (2022).
10. Madani, A. et al.: Large language models generate functional protein sequences across diverse families. Nat Biotechnol 41(8), 1099–1106 (2023). https://doi.org/10.1038/s41587-022-01618-2.
11. Lin, Z. et al.: Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv (2022). https://doi.org/10.1101/2022.07.20.500902.